







ҩ���Ը����ˣ�DILI����Ϊһ�ֳ�����ҩ�ﲻ����Ӧ�����ٴ���ҩ��������Ҫ���Ӻͼ������⡣���Ը�������DILI����ķ�����ʽ���������˻ᷢ����в�����ı����Ի���֢�ι���˥�ߡ�DILI�ķ��������в������ĿǰҲȱ����Ч�����ƴ�ʩ��ҩ�
�ٴ��Ϲ㷺ʹ�õĽ�����ʹҩ�����������ӣ�APAP�����������ÿ��¼��Ը����ˡ���˥�ߣ�����������������APAP�ڸ�ϸ����ͨ��ϸ��ɫ��P450��л����N-���������ǰ���NAPQI��������ǿ�����Կɵ��������幦���ϰ�������Ӧ�����ˣ��Ӷ������ϸ�����������о�����JNK��APAP�յ��ĸ���������ؼ����á�APAP�����ᵼ��JNK���ת�˽�������������յ�������ͨ��ת�䲢���������������ѧ���ܡ����ǣ��ڸ�������JNK���õĵ���������ź�ͨ·�Բ���ȷ��ת¼����Nrf2��ϸ��������ԭ��̬����Ҫ�������ӣ�����һϵ�е�ҩ���лø�Ϳ��������ı����Ч����ҩ���յ��ĸ����ˡ���JNK��Nrf2�������ź�ͨ·�Ƿ���أ���δ������������������������������Ŀ����������ڡ�Hepatology����������Ϊ"JNK Phosphorylates The Neh6 Domain Of Nrf2 And Downregulates Cytoprotective Genes In Acetaminophen�\Induced Liver Injury�������ģ���ʾ����APAP�յ��ĸ������У�Nrf2��JNK���°бꡣ
���о�����APAP�յ��ļ��Ըι���˥�ߵĶ���ģ�ͣ�����APAP��С������м���JNK��ͬʱ��Ҳ����Nrf2���ữ������������ӦԪ����ARE�������Ļ���Nqo1��Gst��3��Gstm1��Gstm5��AKR1C�µ���ͨ��JNK���Ƽ���JNK siRNA����JNK�Ļ���ʱ���ɸ���APAP�յ��ĸζ��ԣ���������Nrf2�����ữ��ARE����������µ�����������������P-JNK��Nrf2��Neh1�ṹ��ֱ������ã���ʹNrf2��Neh6�ṹ���е�SDS1ģ�����ữ���о���Ա���������ͻ����ͻ��ȷ�������ѧ������ȷ��Nrf2��SDS1ģ���е�Ser-335����Ҫ�����ữλ�㣬 JNK��ͨ����λ�����Nrf2�ķ��ػ����⡣�����о���������APAP�յ��ĸ������У�Nrf2��JNK���°б꣬ JNKͨ�����ữNrf2���ƻ���ת¼���ӵ��ص�ϸ������ϵͳ���÷���Ϊ����APAP����ĸ������ṩ���µIJ��ԡ�
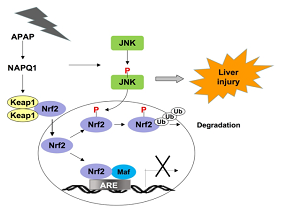
Fig.1 APAP�Ĵ�л��NAQPI����JNK��ͬʱ����Keap1������Nrf2��ˡ�P-JNK���ữNrf2��Neh6�ṹ�������併�⡣�����Nrf2�鵼��ת¼�������ƣ��Ӷ�ϸ���ⶾϵͳ�ͱ���ϵͳ����
��Ϥ����Hepatology����2020��2��17�շ�������Ϊ��Acetaminophen Hepatotoxicity: Strong offense and weakened defense�� �����ۣ��Ը����о������������������
�㽭��ѧ����ҽѧԺ��������ں������Ľ���Ϊ���ĵĹ�ͬͨѶ���ߡ���ʿ������Ƽ��������(�ѱ�ҵ)���ž���Ϊ�����ĵĹ�ͬ��һ���ߡ����о�����˹�����Ȼ��ѧ�����֧�֡�
ԭ�����ӣ�10.1002/hep.31116
Editorial���ӣ�10.1002/hep.31189
