药物性肝损伤(DILI)作为一种常见的药物不良反应,是临床用药过程中需要重视和监测的问题。急性肝损伤是DILI最常见的发病形式,少数病人会发生威胁生命的暴发性或重症肝功能衰竭。DILI的发病机制尚不清楚,目前也缺乏有效的治疗措施和药物。
临床上广泛使用的解热镇痛药对乙酰氨基酚(APAP),过量服用可致急性肝损伤、肝衰竭,甚至死亡。过量的APAP在肝细胞中通过细胞色素P450代谢生成N-乙酰苯醌亚胺(NAPQI),因其强氧化性可导致线粒体功能障碍和氧化应激损伤,从而引起肝细胞死亡。有研究表明JNK在APAP诱导的肝损伤中起关键作用。APAP处理会导致JNK激活并转运进入线粒体进而诱导线粒体通透性转变并抑制线粒体的生物学功能。但是,在肝损伤中JNK作用的底物和下游信号通路仍不明确。转录因子Nrf2是细胞氧化还原稳态的主要调控因子,调控一系列的药物代谢酶和抗氧化蛋白的表达,有效缓解药物诱导的肝损伤。但JNK和Nrf2这两条信号通路是否相关,尚未见报道。最近,王秀君课题组和唐修文课题组联合在《Hepatology》发表了题为"JNK Phosphorylates The Neh6 Domain Of Nrf2 And Downregulates Cytoprotective Genes In Acetaminophen‐Induced Liver Injury”的论文,揭示了在APAP诱导的肝损伤中,Nrf2是JNK的新靶标。
该研究利用APAP诱导的急性肝功能衰竭的动物模型,发现APAP在小鼠肝脏中激活JNK的同时,也引起Nrf2磷酸化,抗氧化剂响应元件(ARE)驱动的基因Nqo1、Gstα3、Gstm1、Gstm5和AKR1C下调。通过JNK抑制剂或JNK siRNA抑制JNK的活性时,可改善APAP诱导的肝毒性,并抑制了Nrf2的磷酸化和ARE驱动基因的下调。生化分析表明,P-JNK与Nrf2的Neh1结构域直接相互作用,并使Nrf2的Neh6结构域中的SDS1模序磷酸化。研究人员用质谱分析和基因点突变等分子生物学方法明确了Nrf2的SDS1模序中的Ser-335是主要的磷酸化位点, JNK可通过该位点调节Nrf2的泛素化降解。这项研究表明,在APAP诱导的肝损伤中,Nrf2是JNK的新靶标, JNK通过磷酸化Nrf2来破坏该转录因子调控的细胞防御系统。该发现为治疗APAP引起的肝损伤提供了新的策略。
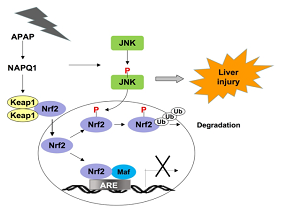
Fig.1 APAP的代谢物NAQPI激活JNK,同时修饰Keap1,导致Nrf2入核。P-JNK磷酸化Nrf2的Neh6结构域并引发其降解。结果,Nrf2介导的转录程序被抑制,从而细胞解毒系统和保护系统受损。
据悉,《Hepatology》在2020年2月17日发表了题为“Acetaminophen Hepatotoxicity: Strong offense and weakened defense” 的评论,对该项研究的意义进行了评述。
浙江大学基础医学院王秀君教授和唐修文教授为该文的共同通讯作者。博士生陈艺萍、刘凯华(已毕业)和张靖文为该论文的共同第一作者。该研究获得了国家自然科学基金的支持。
原文链接:10.1002/hep.31116
Editorial链接:10.1002/hep.31189